
Towards Photonics-Enhanced Molecular Spectroscopy: Label-free Observation of Single Solution-Phase Molecules using Optical Microcavities

Abstract: The vast majority of chemistry and biology occurs in solution, and new label-free analytical techniques that can help resolve solution-phase complexity at the single-molecule level can provide new microscopic perspectives of unprecedented detail. Here, we use the increased light-molecule interactions in high-finesse fiber Fabry-Pérot microcavities to detect individual biomolecules as small as 1.2 kDa (10 amino acids) with signal-to-noise ratios >100, even as the molecules are freely diffusing in solution. Our method delivers 2D intensity and temporal profiles, enabling the distinction of sub-populations in mixed samples. Strikingly, we observe a linear relationship between passage time and molecular radius, unlocking the potential to gather crucial information about diffusion and solution-phase conformation. Furthermore, mixtures of biomolecule isomers of the same molecular weight can also be resolved. Detection is based on a novel molecular velocity filtering and dynamic thermal priming mechanism leveraging both photo-thermal bistability and Pound-Drever-Hall cavity locking. This technology holds broad potential for applications in life and chemical sciences and represents a major advancement in label-free in vitro single-molecule techniques.
 Methanogens are a diverse group of archaea with ancient evolutionary origins. They are found in a wide range of anoxic environments where they carry out a form of anaerobic respiration known as methanogenesis. This process reduces simple oxidized carbon compounds to generate methane as an end product. Another group of archaea related to methanogens carry out the anaerobic oxidation of methane (AOM) and are known as anaerobic methanotrophs (ANME). Methanogens and ANME are both key components in the global carbon cycle and play a central role in controlling atmospheric methane concentrations. Consistent with their anaerobic lifestyles and ancient evolutionary origins, methanogens and ANME contain an abundance of Fe-S cluster proteins. Radical S-adenosylmethionine (SAM) enzymes are [4Fe-4S]-cluster containing enzymes that catalyze a wide variety of difficult biochemical reactions through the generation of a highly reactive 5’-deoxyadenosyl radical. Here, we discuss our recent progress towards uncovering the functions of novel radical SAM enzymes in methanogens and ANME. We identified the missing glutamate 2,3-aminomutase important for salt tolerance in marine organisms as well as characterized the first archaeal methylthiotransferase involved in tRNA modification.
Methanogens are a diverse group of archaea with ancient evolutionary origins. They are found in a wide range of anoxic environments where they carry out a form of anaerobic respiration known as methanogenesis. This process reduces simple oxidized carbon compounds to generate methane as an end product. Another group of archaea related to methanogens carry out the anaerobic oxidation of methane (AOM) and are known as anaerobic methanotrophs (ANME). Methanogens and ANME are both key components in the global carbon cycle and play a central role in controlling atmospheric methane concentrations. Consistent with their anaerobic lifestyles and ancient evolutionary origins, methanogens and ANME contain an abundance of Fe-S cluster proteins. Radical S-adenosylmethionine (SAM) enzymes are [4Fe-4S]-cluster containing enzymes that catalyze a wide variety of difficult biochemical reactions through the generation of a highly reactive 5’-deoxyadenosyl radical. Here, we discuss our recent progress towards uncovering the functions of novel radical SAM enzymes in methanogens and ANME. We identified the missing glutamate 2,3-aminomutase important for salt tolerance in marine organisms as well as characterized the first archaeal methylthiotransferase involved in tRNA modification. 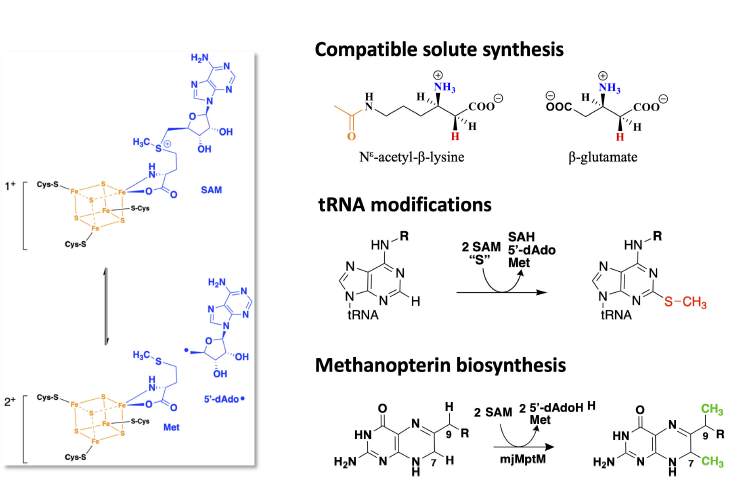
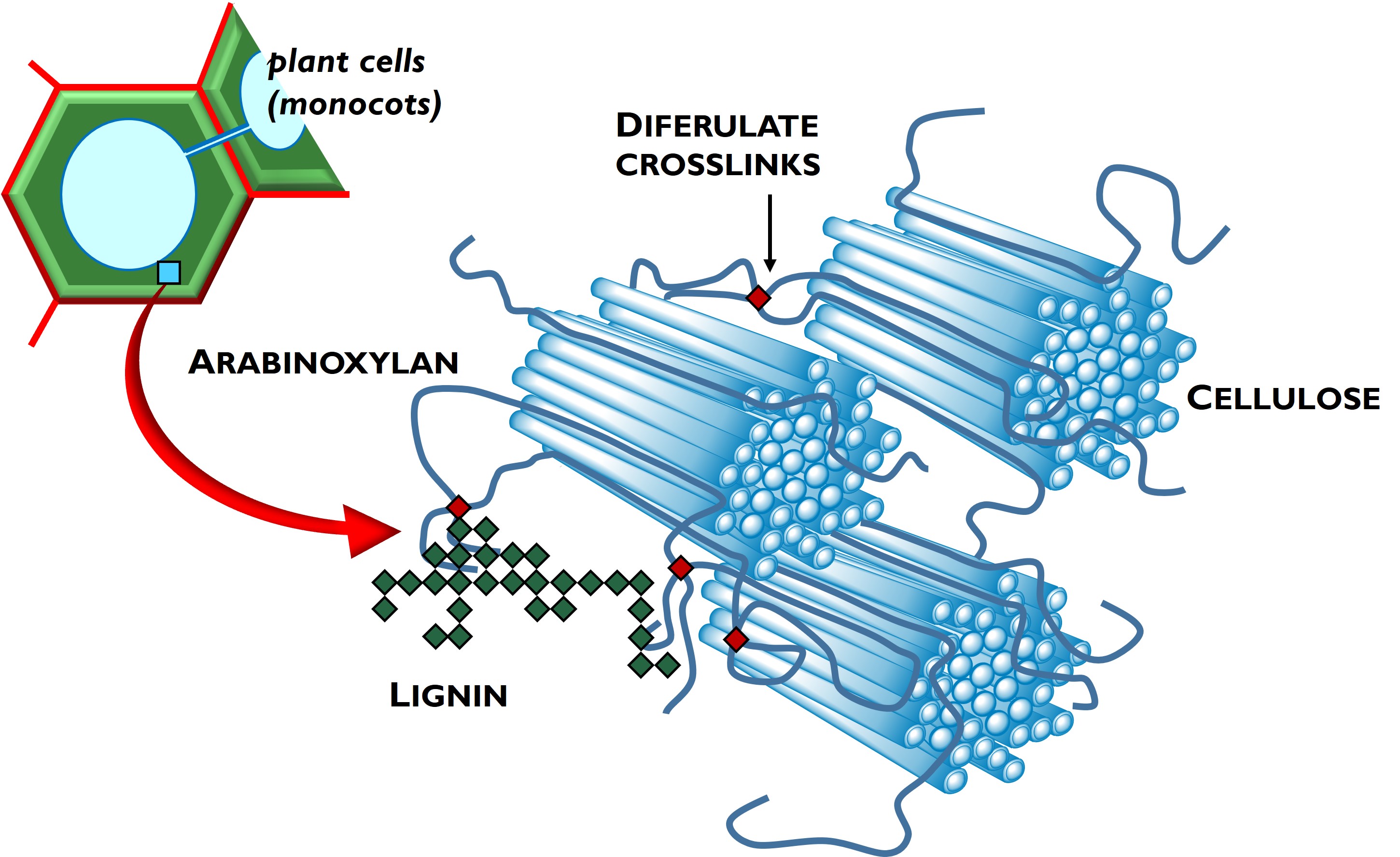 The core areas of Dr. Schendel’s research program at the University of Kentucky are plant cell wall characterization (especially detailed structural analysis of cell wall carbohydrates) and analysis and application of phenolics and other secondary plant metabolites. Strategic collaborations have allowed us to explore applied questions such as ruminant microbe fermentation of cell wall carbohydrates. This seminar will share results from several projects, including our in-depth characterizations of the cell walls of cool-season forages and hempseeds and exploration of their seasonal and species/cultivar variation.
The core areas of Dr. Schendel’s research program at the University of Kentucky are plant cell wall characterization (especially detailed structural analysis of cell wall carbohydrates) and analysis and application of phenolics and other secondary plant metabolites. Strategic collaborations have allowed us to explore applied questions such as ruminant microbe fermentation of cell wall carbohydrates. This seminar will share results from several projects, including our in-depth characterizations of the cell walls of cool-season forages and hempseeds and exploration of their seasonal and species/cultivar variation.  Chamikara Karunasena
Chamikara Karunasena



 Jonathan Rivnay
Jonathan Rivnay


