
Doctoral Exit Seminar: Student Familiarity with the Periodic Table of the Elements: Results from Cued-Recall and Eye-Tracking Assessments on Memory
 Abstract: Learning element symbol-name relationships and the spatial organization of elements on the periodic table is a foundational step in learning chemistry, supporting later understanding of chemical formulas, equations, bonding and stoichiometry. Although students often rely on memorization strategies to learn periodic table content, this task is challenging due to the large number of elements and the apparent ambiguity in matching some element symbols to their names.
Abstract: Learning element symbol-name relationships and the spatial organization of elements on the periodic table is a foundational step in learning chemistry, supporting later understanding of chemical formulas, equations, bonding and stoichiometry. Although students often rely on memorization strategies to learn periodic table content, this task is challenging due to the large number of elements and the apparent ambiguity in matching some element symbols to their names.
This study explores students’ recall of element names when given element symbols as cues and their knowledge of element locations on the periodic table when given element names as cues. The study also examines how these two pieces vary across course groups. Seven instructional groups representing increasing levels of chemistry coursework were examined using two complementary but distinct experimental approaches: a survey-based cued-recall task assessing percentage-correct symbol-name recall, and a separate eye-tracking experiment assessing how students visually searched for selected elements on the periodic table.
The overall results with all elements considered together reveal that recall of element symbol-name relationships and visual search efficiency improve with increasing chemistry exposure, but do not follow a strictly stepwise progression. Element-specific analyses reveal that recall of symbol-name relationships developed unevenly across the periodic table:
- Some elements consolidate early.
- Others strengthen gradually with increased exposure to chemistry.
- A subset remain weakly recalled even among upper-level undergraduate and graduate students.
Visual search efficiency follows a similar pattern, shifting from exploratory search to shorter, more focused search paths in advanced course groups. Recall and visual search efficiency were similar but not equivalent. In some cases, elements were located efficiently despite weak or absent recall, whereas in others elements recalled were associated with inefficient search behavior.
These findings indicate that symbol-name knowledge and spatial knowledge of element locations constitute distinct but interacting memory components. At the upper levels of instruction, there seems to be an integration of these components, enabling students to use the periodic table more effectively as a structured representation rather than relying solely on element symbol-name associations.
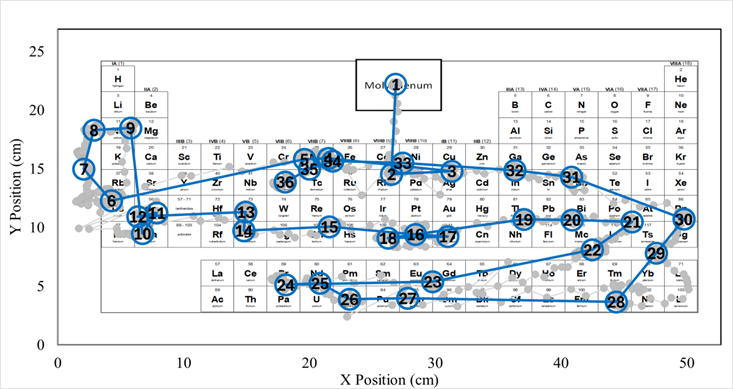
KEYWORDS: Periodic table, symbol–name relationalships, visual search, search behavior, consolidation, integration
 Abstract:
Abstract: 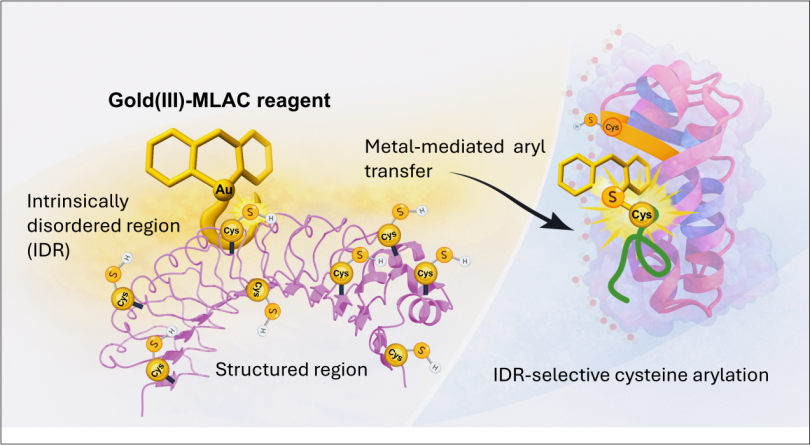
 Abstract:
Abstract: 
 Abstract: Cancer cells have developed uncanny strategies to evade the effectiveness of anticancer therapies and immune destruction by modulating their energy metabolism to a pro-survival state. This altered metabolism supports their proliferation and enables niches to thrive even in the presence of unfavorable conditions.
Abstract: Cancer cells have developed uncanny strategies to evade the effectiveness of anticancer therapies and immune destruction by modulating their energy metabolism to a pro-survival state. This altered metabolism supports their proliferation and enables niches to thrive even in the presence of unfavorable conditions. 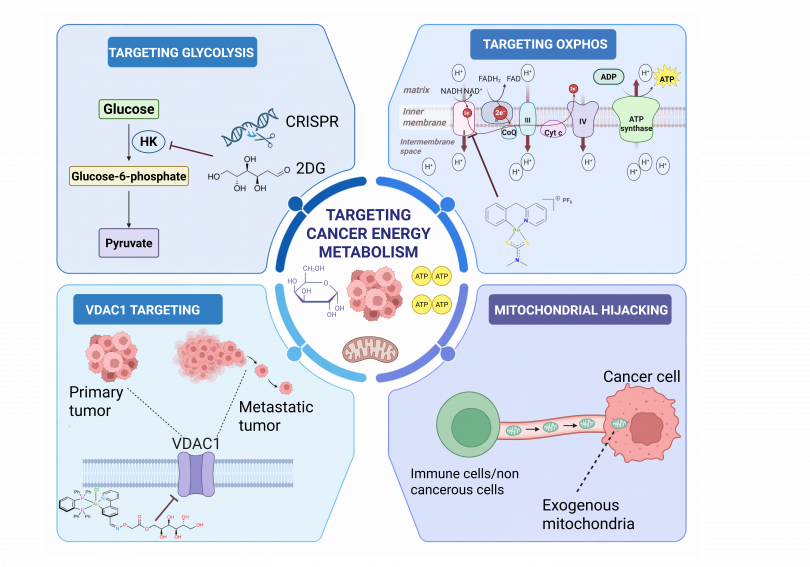
 Abstract: The endoplasmic reticulum (ER) chaperone, glucose-regulated protein (GRP78)/binding immunoglobulin protein (BiP)/HSPA5, is a master regulator of Proteostasis, regulating protein folding, the Unfolded Protein Response (UPR) and Endoplasmic Reticulum-associated degradation (ERAD). GRP78 is often overexpressed in many cancers, and this vulnerability has been therapeutically targeted, but therapeutic success has been hampered by resistance and immunosuppression. Despite the availability of a few inhibitors of GRP78, none have achieved clinical approval, highlighting a critical need for new therapeutic strategies.
Abstract: The endoplasmic reticulum (ER) chaperone, glucose-regulated protein (GRP78)/binding immunoglobulin protein (BiP)/HSPA5, is a master regulator of Proteostasis, regulating protein folding, the Unfolded Protein Response (UPR) and Endoplasmic Reticulum-associated degradation (ERAD). GRP78 is often overexpressed in many cancers, and this vulnerability has been therapeutically targeted, but therapeutic success has been hampered by resistance and immunosuppression. Despite the availability of a few inhibitors of GRP78, none have achieved clinical approval, highlighting a critical need for new therapeutic strategies. 